
 返回
返回
系列①—⑤,我们分别解读了四种分类调整情形和三条快速通道。
今天进入系列⑥——也是本系列的最后一篇:《医疗器械分类目录动态调整工作程序》。
如果说前面的篇章是讲“怎么应对分类调整”,那么这一篇就是讲 “分类目录本身怎么变化” 。这门“背后的工作程序”,决定了你未来面对的分类规则是如何制定的。
2026年6月1日,国家药监局发布2026年第53号公告,正式印发修订后的《医疗器械分类目录动态调整工作程序》,同步废止2021年第60号公告版本。这意味着医疗器械分类目录正式进入“常态化动态调整”时代。
这篇文章,我们把这套工作机制的核心内容讲清楚。👇
医疗器械行业处在不断创新发展的动态变革之中,新型器械、创新应用不断涌现,既往分类目录更新的节奏和时效性面临挑战。《医疗器械监督管理条例》明确规定,国家药监局需根据医疗器械生产、经营、使用情况,及时对风险变化进行分析、评价,对分类目录进行动态调整。
动态调整工作遵循以下原则:
根据医疗器械风险变化情况
遵循符合最新科学认知
立足监管实际、借鉴国际经验
支持鼓励创新、推动高质量发展
修订后的《动态调整工作程序》构建起“风险导向、动态适配、高效透明”的分类调整体系,覆盖医疗器械及体外诊断试剂分类子目录调整工作。
《动态调整工作程序》第四条明确了分类目录调整的五种情形:
增加、删除子目录(结构性调整)
调整一级产品类别、二级产品类别
调整已列入目录产品的管理类别、管理属性,或在目录中明确相关产品的管理属性和管理类别
修订产品描述、预期用途等内容
调整品名举例内容,如增补有代表性的产品、删除不再作为医疗器械管理的产品
💡 这五种情形基本囊括了目录中与产品管理类别及属性有关的全部内容,确保调整工作有章可循。
动态调整不是监管部门“关门开会”,而是开放给社会各界参与的。根据程序第六条和第七条:
面向所有相关方:
境内医疗器械注册人、备案人
医疗器械生产、经营和使用单位
设区的市级负责药品监督管理的部门
国家药监局相关部门、省级药品监督管理部门
医疗器械相关学会、协会等社会团体
医疗器械分类技术委员会委员
操作路径:
境内申请人向所在地省级药品监督管理部门提出建议
境外申请人可委托境内代理人,向其代理人所在地省级药监部门提出
省级药监部门初审后,经评估建议调整的,报器械标管中心
国家药监局相关部门、协会、技术委员会委员等可直接向器械标管中心提出
💡 如果企业发现自己的产品分类不合理、风险程度发生变化,或者有新产品需要明确管理类别,都可以走这个渠道提出调整建议。分类目录动态调整将会成为常态工作,需要社会各界积极参与。
根据程序第八条,调整建议原则上应当通过器械标管中心分类界定信息系统提交,相关材料主要包括:
拟调整的内容和理由
拟调整产品国内外管理属性、类别和产业现状,以及在我国注册/备案情况
拟调整产品主要风险点及风险变化情况
拟调整产品技术特点、与已上市同类产品的比较和临床使用情况
拟调整产品不良事件和上市后监管有关情况(如适用)
拟调整产品过渡期建议
其他与目录调整有关的资料
💡 实操提示:材料越充分,论证越有说服力。尤其是产品风险分析、国内外同类产品对比、过渡期建议等,都是影响调整决策的关键因素。
分工明确,各司其职:
器械标管中心:定期梳理汇总分类界定结果,形成初步调整意见;组织研究;将拟调整内容在其网站向社会公开征求意见
医疗器械分类技术委员会执委会:审议子目录整体拟调整意见(如增加/删除子目录)
医疗器械分类技术委员会专业组:审议具体品类调整等其他情形
征求意见环节:一般不少于30个自然日;对于监管急需的产品,可缩短为不少于7个自然日。
💡 这套机制既保证了决策的专业性(技术委员会把关),又保证了过程的公开性(征求意见、公示),同时也兼顾了效率(监管急需可缩短期限)。
作为系列前五篇中反复引用的条款,《动态调整工作程序》第十条是过渡期设置的核心法规依据。
根据第十条(一):由不作为医疗器械管理调整为第二类或第三类,或由低类别调整为高类别的,一般设置2-3年过渡期。不须开展临床试验的过渡期一般设为2年,须开展临床试验的设为3年。
根据第十条(二):对于注册用时较长的情形(如医疗器械调整为药品管理),可设置3年以上过渡期,原则上最长不超过5年。
根据第十条(三):产品风险高、经论证需要立即规范的,可不设过渡期。
根据第十条(四):已有产品上市但尚未纳入分类目录、管理属性已有共识的,纳入目录时可不设过渡期。
💡 关于何时设置过渡期、过渡期设置多久的决定程序,本程序第十条也明确了要求:器械标管中心应当综合考虑产品风险、监管风险、产业发展、临床应用需求等因素,遵循科学规律,充分研究和论证,必要时组织调研,提出是否设置过渡期及过渡期设置时限。
本次修订对2021年第60号公告版本作出四大调整:
① 更新法规依据、扩大目录范围
增加《体外诊断试剂分类规则》作为动态调整法规依据,增加《体外诊断试剂注册与备案管理办法》作为动态调整后注册备案事项的法规依据,将《体外诊断试剂分类目录》纳入适用目录范围,实现医疗器械与体外诊断试剂分类调整规则统一衔接。
② 明确过渡期设置原则
即上文详细拆解的第十条内容。
③ 细化各环节要求
如征求意见环节,监管急需产品可适当缩短公开征求意见期限,提升工作效率。
④ 增加各方职责要求
明确器械标管中心、分类技术委员会执委会、专业组的具体职责分工。

📢 系列回顾
本系列共六篇至此告一段落:
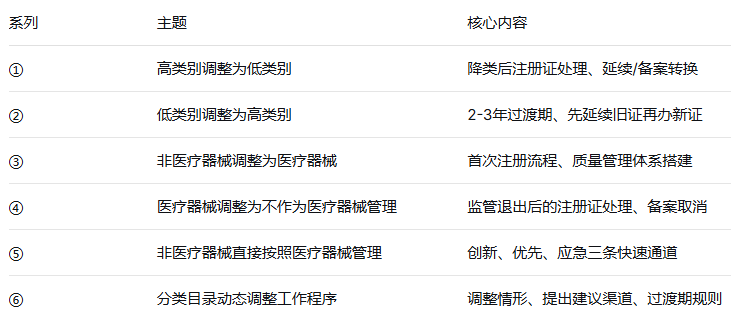
分类调整不是“纸上谈兵”,每一类产品都有对应的行动方案。希望这个系列能帮助企业在面对分类调整时——心中有数,手中有策。🔚
关于我们
2026世界杯赛程药械咨询技术服务集团——国内领先的医疗器械一站式注册认证咨询与软件开发管理综合性服务集团,成立于2007年。为国内外医疗器械企业提供从产品立项、设计开发、厂房规划、检验检测、临床评价、质量管理体系到注册认证的“全方位、一站式”解决方案。
✨ 尤其擅长美容类医疗器械(射频、激光、强脉冲光、水光针、胶原蛋白、透明质酸钠等)国内注册及FDA、CE等国际认证。
📞 欢迎联系2026世界杯赛程专业团队!
📢 本文根据国家药监局2026年第53号公告附件《医疗器械分类目录动态调整工作程序》、2026年第52号和第53号公告官方解读等官方文件整理,仅供参考。具体执行请以官方文件及药监部门解释为准。

